本项目主要用于对扩增子测序数据经qiime2分析产生的ASV丰度数据及其分类注释结果进行下游分析和结果可视化。
[TOC]
01 ASV丰度数据过滤 [01Amplicon_rawdate_filter_stats.R]
(包括非细菌、线粒体、叶绿体及样本重复性差的ASV过滤[min_reads >=10, min_samples >=3])
02 稀疏曲线 (rarecurve)数据分析及可视化 [02Amplicon_rarecurve.R]
03 数据归一化 (Flatten or CSS) [03Amplicon_Flatten_or_CSS.R]
04 alpha多样性分析 [04Amplicon_alpha_diversity.R]
05 beta多样性分析 [05Amplicon_beta_diversity.R]
06 Taxonomy表格整理 [06Split_taxonomy.R]
07 物种组成 (各分类水平相对丰度表格生成) [07Amplicon_relative_abundance.R]
其他:
ASV丰度数据拆分 [Split_ASV_by_metadata.R]
分组信息表格拆分 [Split_metadata.R]
alpha多样性组间差异分析 [Alpha_diversity_diff_factor1.R]
[Alpha_diversity_diff_factor2.R]
本流程依赖的 R 包可在 R version 4.4.2 下正常运行。
./AmpliconR/
├── ReadMe.md
├── ReadMe.pdf
├── AmpliconR_pipeline.R
├── asv.table.txt
├── asv.taxa.txt
├── sample_info.txt
├── Rscript/
│ ├── 01Amplicon_rawdate_filter_stats.R
│ ├── 02Amplicon_rarecurve.R
│ ├── 03Amplicon_Flatten_or_CSS.R
│ ├── 04Amplicon_alpha_diversity_calculate.R
│ ├── 05Amplicon_beta_diversity.R
│ ├── 06Split_taxonomy.R
│ ├── 07Amplicon_relative_abundance.R
│ ├── Split_ASV_by_metadata.R
│ ├── Split_metadata.R
│ ├── Alpha_diversity_diff_factor1.R
│ └── Alpha_diversity_diff_factor2.R
├── results/
└── PlotScript/
├── 02Amplicon_rarecurve_plot.R
├── 04Amplicon_alpha_factor1_plot.R
├── 04Amplicon_alpha_factor2_plot.R
└── 05Amplicon_beta_plot.R
Github下载链接:https://github.com/shuailuo2021/AmpliconR.git
setwd("yourpath/AmpliconR/")
# 新建 results 文件夹
new_folder <- "results"
if (!dir.exists(new_folder)) {
dir.create(new_folder)
} else {
print("文件夹已存在。")
}
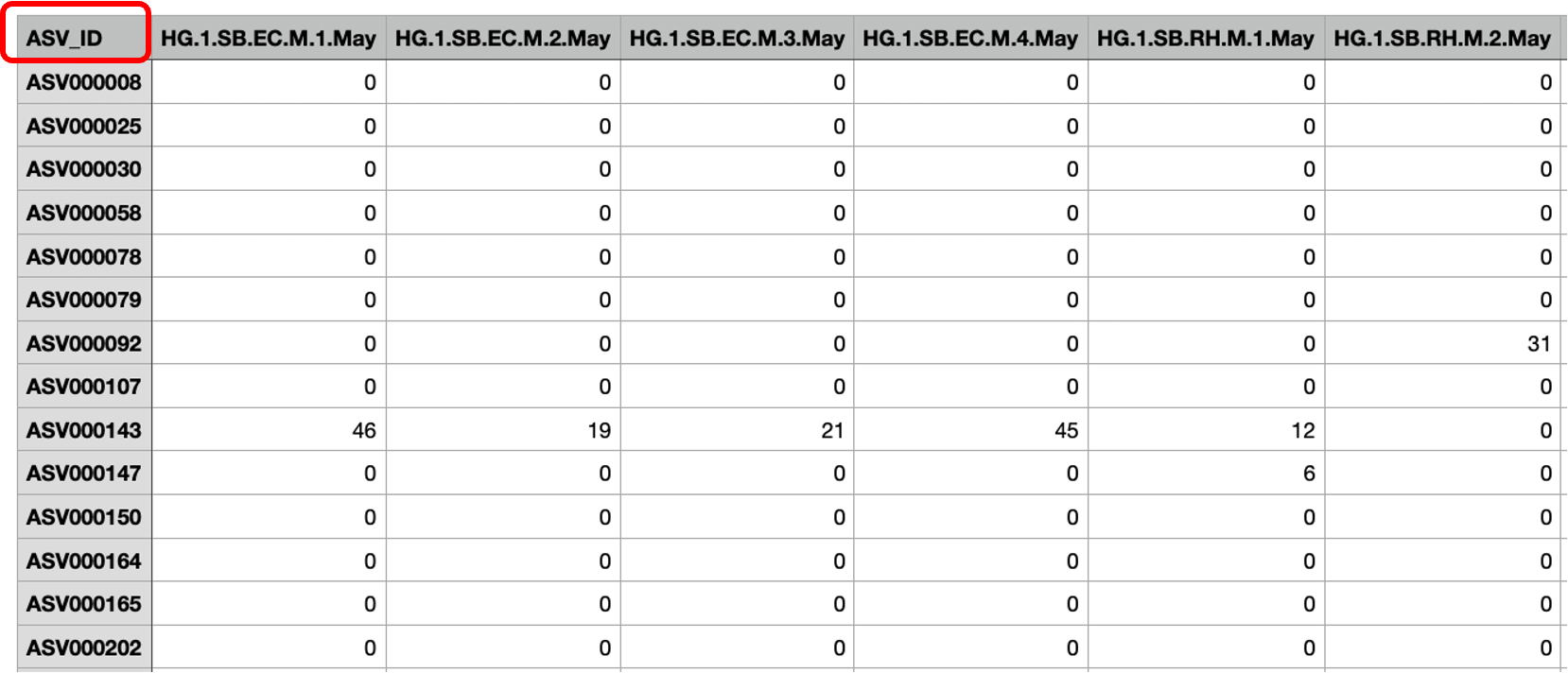
注:ASV丰度数据表每行为ASV序号名,每列为样本编号。
要求ASV丰度数据表的第一列列名为“ASV_ID”,与以下分类注释表的第一列列名一致
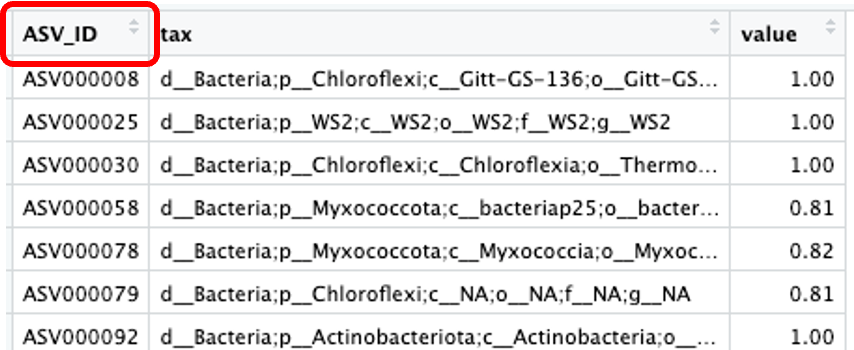
注:ASV分类注释表每行为ASV序号名及对应的分类注释信息和相似度。
要求ASV分类注释表的第一列列名为“ASV_ID”,与以上ASV丰度数据表的第一列列名一致
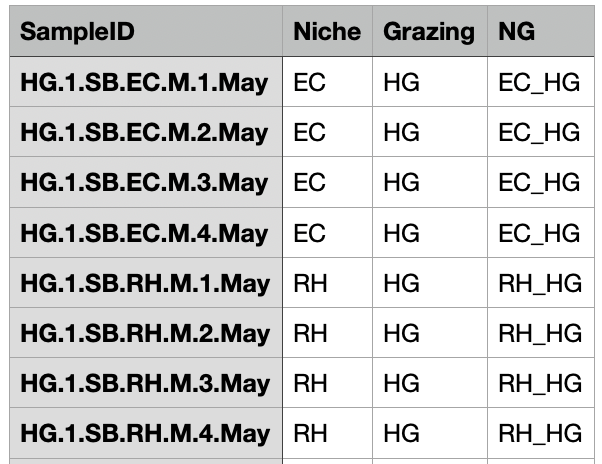
注:样本分组信息表每行为样本名称及对应的分组,具体根据自身情况而定。
要求样本分组信息表的第一列列名必须为“SampleID”。
调用相应分析脚本 source(“./scripts/***.R”)
具体使用方法见 ./AmpliconR_pipeline.R
# 调用脚本
source("./Rscript/01Amplicon_rawdate_filter_stats.R")
result <- process_asv_data_with_stats(
asv_file_path = “./asv.table.txt",
tax_file_path = "./asv.taxa.txt",
min_reads = 10,
min_samples = 3,
output_dir = "./results/"
)
该脚本主要用于对原始ASV数据表进行过滤,包括:
- 去除非细菌ASV(保留分类注释含"d__Bacteria"的ASV)
- 去除分类注释含线粒体(Mitochondria)、叶绿体(Chloroplast)的ASV
- 保留至少在min_samples(3)个样本中reads数达到min_reads(10)的ASV
结果文件:
./results/ASV_keep.txt过滤后ASV table./results/taxonomy_keep.txt过滤后taxonomy./results/01processing_stats.txt过滤过程统计信息

# 调用脚本
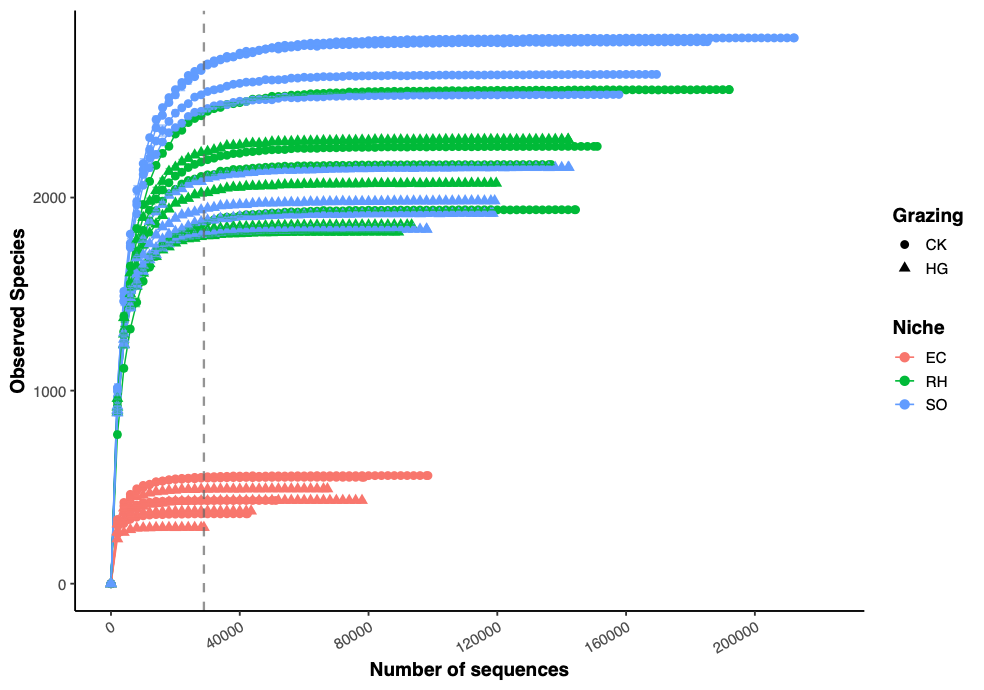
source("./scripts/02Amplicon_rarecurve.R")
# 自动调整横坐标范围
rare_plot <- plot_rare_curve(
ASV_table_path = "./results/ASV_keep.txt",
sample_info_path = "./sample_info.txt",
color_var = "Niche",
group_var = "Grazing",
shape_var = "Grazing",
output_path = "./results/02rarecurve_auto_range.pdf"
)
# 手动指定横坐标范围
rare_plot_custom <- plot_rare_curve(
ASV_table_path = "./results/ASV_keep.txt",
sample_info_path = "sample_info.txt",
group_var = "Grazing",
color_var = "Niche",
shape_var = "Grazing",
x_breaks = seq(0, 250000, by = 30000),
output_path = "./results/02rarecurve_custom_range.pdf"
)
以上可通过默认设置和手动指定横坐标范围,二选一
根据稀疏曲线结果可用于确定数据归一化方法和抽平值
结果文件:
02plot_richness_stat_meta.txt结果文件可用于自定义绘图,可参考./PlotScript/02Amplicon_rarecurve_plot.R02rarecurve_auto_range.pdf默认输出图片
# 调用脚本
source("./scripts/03Amplicon_Flatten_or_CSS.R")
# flatten归一化示例
flatten_result <- normalize_flatten(
input_file = "./results/ASV_keep.txt",
output_file = "./results/ASV_keep_norm.txt",
min_reads = 10,
min_samples = 3,
seed = 123
)
# 查看处理过程信息
attributes(flatten_result)$processing_info

结果文件:
./results/ASV_keep_norm.txt
# 调用脚本
source("./scripts/03Amplicon_Flatten_or_CSS.R")
css_result <- normalize_CSS(
input_file = "./results/ASV_keep.txt",
output_file = "./results/ASV_keep_norm_CSS.txt",
min_reads = 10,
min_samples = 3
)
# 查看处理过程信息
attributes(css_result)$processing_info
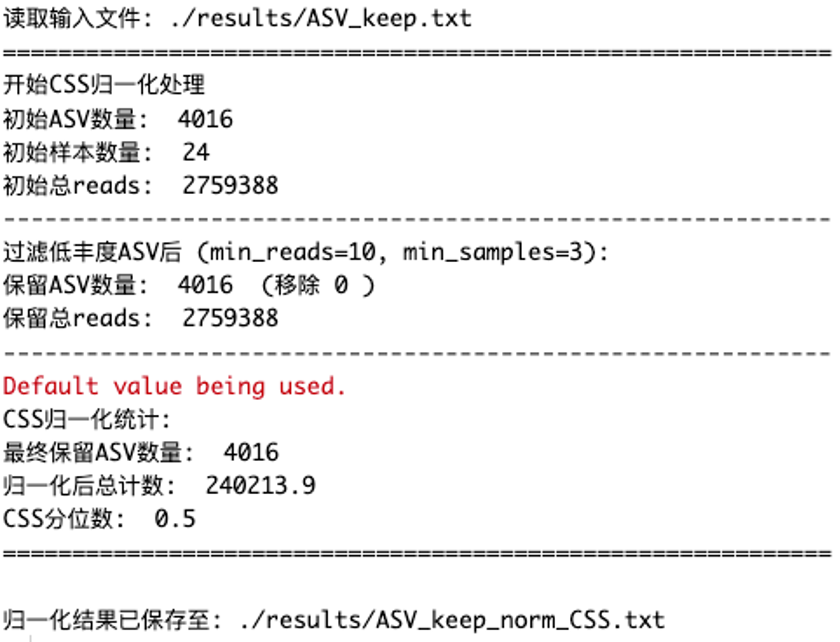
结果文件:
./results/ASV_keep_norm_CSS.txt
# 调用脚本
source("./scripts/04Amplicon_alpha_diversity.R")
calculate_alpha_diversity(
sample_info_file = "sample_info.txt",
asv_norm_file = "./results/ASV_keep_norm.txt",
output_file = "./results/04alpha_diversity.txt"
)
结果文件:
./results/04alpha_diversity.txt
# 调用脚本
source("./scripts/05Amplicon_beta_diversity.R")
beta_diversity_analysis(
norm_asv_path = "./results/ASV_keep_norm.txt",
sample_info_path = "sample_info.txt",
output_dir = “./results/beta_all/"
)
结果文件:
./results/beta_all/05beta_pcoa.txtbeta多样性结果文件./results/beta_all/05pcoa_eigenvalues.txt特征值解释度
结果文件可用于自定义绘图,可参考./PlotScript/05Amplicon_beta_plot.R
# 调用脚本
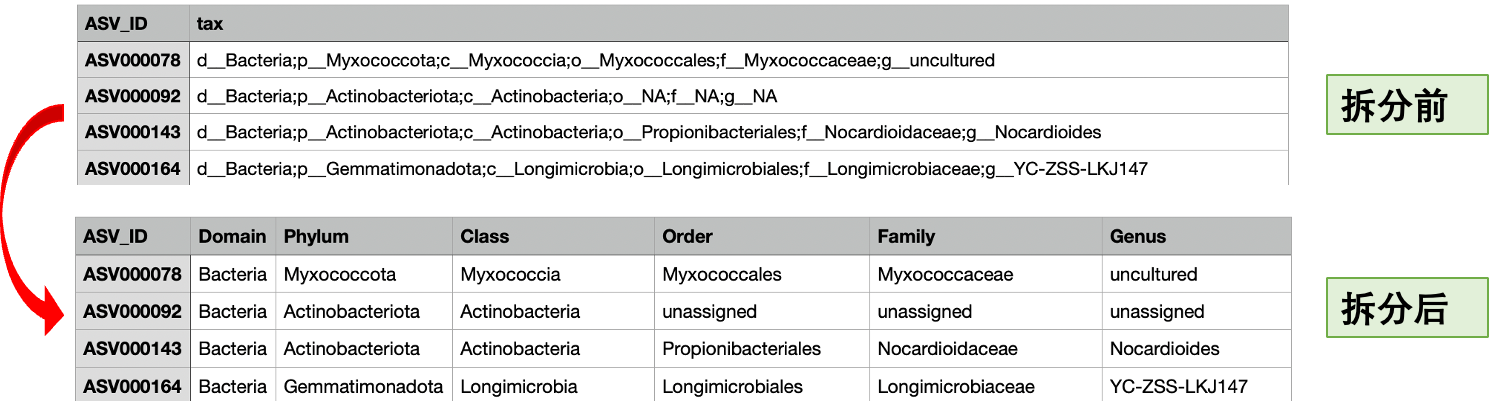
source("./scripts/06Split_taxonomy.R")
# 保存文件并返回数据框
result_df <- process_taxonomy_table(
input_file = "./results/taxonomy_keep.txt",
output_file = "./results/taxonomy_split.txt"
)
结果文件./results/taxonomy_split.txt
# 调用脚本
source("./scripts/07Amplicon_relative_abundance.R")
# 完整用法(保存文件到指定目录)
result <- calculate_relative_abundance(
taxonomy_file = "./results/taxonomy_split.txt",
abundance_file = "./results/ASV_keep_norm.txt",
output_dir = "./results/06relative_abundance"
)
# 从RDS文件重新加载结果
RA_results <- readRDS("./results/06relative_abundance/Relative_Abundance_Results.rds")
Phylum_RA_results <- RA_results$Phylum
结果文件:

# 调用脚本
source("./scripts/Split_ASV_by_metadata.R")
# Split by Niche
split_asv_by_metadata(
meta_file = "./sample_info.txt",
asv_file = "./results/ASV_keep.txt",
group_column = "Niche"
)
如根据生态位拆分ASV table的结果文件:
./AmpliconR/results/ASV_keep_Niche
├── ASV_keep_SO.txt
├── ASV_keep_RH.txt
└── ASV_keep_EC.txt
# 调用脚本
source("./scripts/Split_metadata.R")
result <- split_metadata(
input_file = "./sample_info.txt",
output_file = "./sample_info_split.rds",
group_vars <- c("Niche","Grazing","NG")
)
根据分组变量拆分的结果文件:./sample_info_split.rds
# 从RDS文件读取结果
MetaSplit <- readRDS("./sample_info_split.rds")
# 提取特定分组数据
meta_SO <- MetaSplit$Niche$SO
meta_CK <- MetaSplit$Grazing$CK
meta_SO_CK <- MetaSplit$NG$SO_CK
# 调用脚本
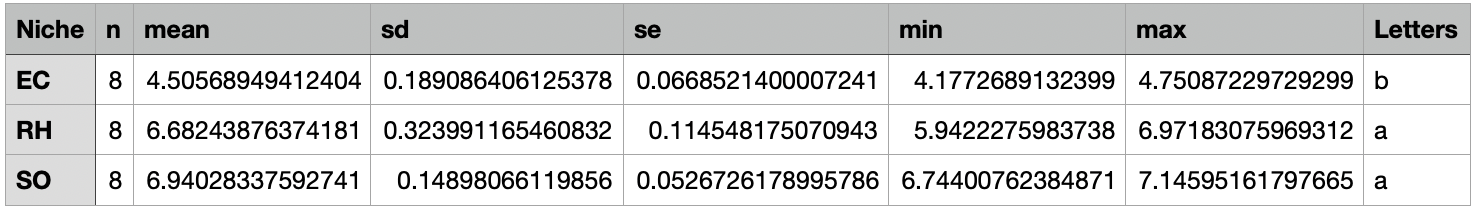
source("./scripts/Alpha_diversity_diff_factor1.R")
# Shannon
result <- alpha_diversity_stats(
input_path = "./results/04alpha_diversity.txt",
output_path = "./results/04alpha_stats_Niche_Shannon.txt",
group_column = “Niche”, # 分组列名
value_column = "Shannon" # 数值列名
)
# Chao1
result <- alpha_diversity_stats(
input_path = "./results/04alpha_diversity.txt",
output_path = "./results/04alpha_stats_Niche_Chao1.txt",
group_column = "Niche", # 分组列名
value_column = "S.chao1" # 数值列名
)
结果文件:
./results/04alpha_stats_Niche_Shannon.txtShannon组间差异分析结果./results/beta_all/04alpha_stats_Niche_Chao1.txtChao1组间差异分析结果
结果文件可用于自定义绘图,可参考./PlotScript/04Amplicon_alpha_factor1_plot.R
# 调用脚本
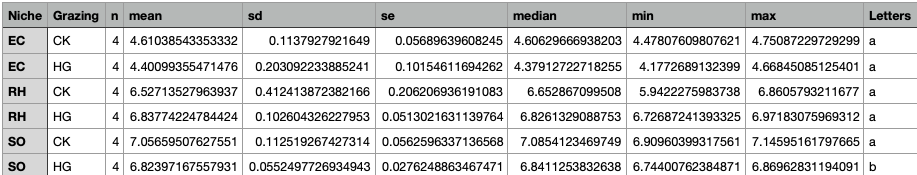
source("./scripts/Alpha_diversity_diff_factor2.R")
# Shannon
result <- kruskal_group_comparison(
input_file = "./results/04alpha_diversity.txt",
output_file = "./results/04alpha_kruskal_results_Shannon.txt",
group_var = "Niche",
compare_var = "Grazing",
value_var = "Shannon"
)
# Chao1
result <- kruskal_group_comparison(
input_file = "./results/04alpha_diversity.txt",
output_file = "./results/04alpha_kruskal_results_Chao1.txt",
group_var = "Niche",
compare_var = "Grazing",
value_var = "S.chao1"
)
结果文件:
./results/04alpha_kruskal_results_Shannon.txtShannon组间差异分析结果./results/04alpha_kruskal_results_Chao1.txtChao1组间差异分析结果
结果文件可用于自定义绘图,可参考./PlotScript/04Amplicon_alpha_factor2_plot.R